L’incremento dello spazio morto riduce la ventilazione alveolare in assenza di un adeguato incremento della ventilazione minuto. Quando questo accade, aumenta la PaCO2. L’ipercapnia però non è causata solamente dall’aumento dello spazio morto, ma può anche essere secondaria ad una riduzione della ventilazione minuto (vedi anche il post del 17/02/2018).
Il post di oggi mette a fuoco questa articolata relazione tra spazio morto, ipoventilazione ed ipercapnia e propone due semplici surrogati di spazio morto (Ventilazione Minuto corretta e Ventilatory Ratio).
Ventilazione minuto, ventilazione alveolare, spazio morto e PaCO2.
Siamo abituati a misurare la ventilazione del paziente come ventilazione minuto (VE), ossia il prodotto tra volume corrente (VT) e frequenza respiratoria (FR), che quantifica quanti litri di aria entrano ed escono nelle vie aeree in 1 minuto. Un soggetto sano, come ci insegna la fisiologia, inspira un VT circa 0.5 L circa 12 volte al minuto. Questo significa che la sua VE è 6 L/min.
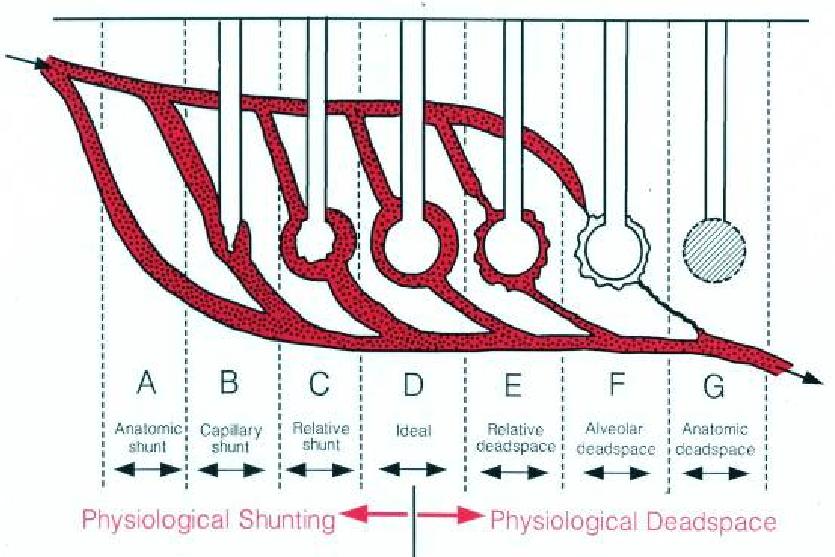
La PaCO2 dipende da quella parte di VE che definiamo ventilazione alveolare (VA). La ventilazione dello spazio morto (VD) è la differenza tra VE e VA, ossia di quella parte di VE che non partecipa allo scambio gassoso. In fisiologia lo spazio morto si identifica solamente con il volume di aria che si ferma nelle vie aeree (spazio morto anatomico), circa 150 mL.

La VA è quindi calcolata come:
VA = (VT - VD) ∙ FR
In un soggetto maschio sano, che chiameremo con originalità Mario Rossi, utilizzando i suddetti dati possiamo calcolare la VA:
VA = (0.5 L - 0.15 L) ∙ 12/min = 4,2 L/min.
La PaCO2 è inversamente proporzionale alla VA:
PaCO2 ~ 1/VA
e Mario Rossi ha la sua normale PaCO2 di 40 mmHg con una VA di 4,2 L/min, che corrisponde ad una VE di 6 L/min.
L’ipoventilazione.
Le formule precedenti ci dicono che se Mario Rossi dimezza la frequenza respiratoria (da 12 a 6/min), la VA diventà la metà, e pertanto raddoppia la PaCO2 a 80 mmHg.
E’ una condizione facilissima da gestire: per tornare ad una PaCO2 di 40 mmHg è sufficiente iniziare la ventilazione meccanica, impostando una frequenza respiratoria di 12/min ed un volume corrente di 500 mL, cosa che tutti gli anestesisti fanno ogni volta in cui inducono una anestesia generale.
L’ipoventilazione è l’ultimo dei problemi per chi sa gestire la ventilazione meccanica, a meno che non riesca ad intubare o le vie aeree siano ostruite…
Lo spazio morto.
Ipotizziamo che a Mario Rossi venga una ARDS e sia ventilato con volume corrente di 430 ml e frequenza respiratoria 28/min. Con questa impostazione la VE è 12 L/min (il doppio del normale) e, se lo spazio morto rimanesse a 150 ml, la VA sarebbe 7.8 L/min (quasi il doppio del normale) e la PaCO2 di conseguenza sarebbe quasi la metà del normale (poco più di 20 mmHg).
Ma sappiamo bene che la realtà è ben diversa: quando Mario Rossi ha una ARDS, potrebbe essere ipercapnico nonostante l’incremento di VA e VE.
Nella ARDS aumenta lo spazio morto alveolare (che è trascurabile in fisiologia), che rappresenta quel volume d’aria che non partecipa allo scambio gassoso pur essendo negli alveoli. Lo spazio morto alveolare si genera se esistono superfici alveolari che non ricevono la perfusione dei capillari polmonari oppure se i polmoni sono caratterizzati da aumento ed eterogeneità del rapporto ventilazione/perfusione. Nella ARDS lo spazio morto fisiologico, cioè la somma di spazio morto anatomico ed alveolare, può anche arrivare al 80% del volume corrente. Con questo valore si spazio morto, è possibile calcolare che nel nostro Mario Rossi vi sia una PaCO2 di 70 mmHg nonostante l’iperventilazione.
A differenza della condizione di ipoventilazione, l’ipercapnia dovuta ad incremento dello spazio morto è particolarmente difficile da risolvere, perché richiederebbe un notevole incremento della VE, attraverso l’aumento di volume corrente e frequenza respiratoria, scelte che rischiano di facilitare il Ventilator-Induced Lung Injury: un correttivo ben peggiore del problema (presunto) che si vuole risolvere.
In questo caso è meglio accettare l’ipercapnia, che è un marker di gravità più che un problema, ed aspettare che si risolva seguendo la guarigione del paziente.
I surrogati dello spazio morto.
Lo spazio morto è la variabile maggiormente associata alla mortalità nei pazienti con ARDS. Conoscerlo può avere un valore prognostico, può consentire di comprendere correttamente il significato di iperventilazione ed ipercapnia, può essere utile nel valutare l’evoluzione della ARDS quando variano sia la VE che il valore di PaCO2. Lo spazio morto, nella valutazione dei pazienti con ARDS, dovrebbe perlomeno affiancare il ben noto PaO2/FIO2, che come sappiamo è soggetto a molte limitazioni (vedi post del 29/01/2017).
Il calcolo dello spazio morto richiede però la misurazione della pressione parziale media di CO2 nell’espirato, dato che spesso non è disponibile perchè richiede strumenti appropriati come capnometria volumetrica o calorimetria indiretta.
Abbiamo però la possibilità di utilizzare due surrogati della misura dello spazio morto: VE corretta (VEcorr) e Ventilatory Ratio. Pur avendo entrambi questi indici il limite di utilizzare il VE come indicatore di VA (abbiamo visto che non è la stessa cosa), si sono dimostrati associati alla prognosi nei pazienti con ARDS, similmente allo spazio morto. Possono pertanto essere utili surrogati dello spazio morto nella pratica clinica.
La ventilazione minuto corretta (VEcorr).
La VEcorr indica quanto dovrebbe essere la VE per ottenere la normocapnia.
E’ molto facile da calcolare:
VEcorr = VE ∙ PaCO2/(PaCO2 fisiologica),
dove la PaCO2 fisiologica è stimata in 40 mmHg.
Se un soggetto è normocapnico, VE e VEcorr coincidono.
Calcoliamo ora la VEcorr di Mario Rossi quando ipoventila e quando ha l’ARDS, come descritto nei due esempi precedenti.
Quando Mario Rossi ipoventila:
VEcorr = 3 L/min ∙ 80 mmHg/40 mmHg = 6 L/min
In questo esempio la VEcorr è uguale al VE "da libro di fisiologia", e possiamo concludere che lo spazio morto è assente. Una buona notizia per il paziente (basso spazio morto si associa a minore probabilità di morte) ed una condizione molto facile da correggere.
Nei pazienti con ARDS, la VEcorr si associa ad incremento di mortalità quando è superiore a 13 L/min.
Calcoliamo VEcorr nel nostro Mario Rossi con l’ARDS:
VEcorr = 12 L/min ∙ 70 mmHg/40 mmHg = 21 L/min
Questo significa che Mario Rossi dovrebbe avere una VE di 21 L/min per ottenere una PaCO2 di 40 mmHg: un valore elevatissimo che è indice di un grave aumento dello spazio morto. Una cattiva notizia per Mario Rossi ed una condizione che ci deve indurre a tollerare l’ipercapnia come male minore rispetto ad un’escalation della ventilazione.
Ventilatory Ratio.
Il Ventilatory Ratio indica di quante volte deve essere aumentata la VE, rispetto al valore ideale, per ottenere la normocapnia. A differenza di VEcorr, il Ventilatory Ratio è un numero adimensionale che tiene conto anche del livello di VE ideale, che a sua volta dipende dall’altezza del paziente. Si ritiene che il VE ideale sia 0.1 L/min per kg di peso ideale. Una persona di bassa statura, con peso ideale di 48 Kg, ha un VE ideale di 4.8 L/min, ben diverso dagli 8 L/min che rappresentano il VE ideale di persona alta 185 cm e peso ideale di 80 kg.
Il Ventilatory Ratio si calcola come:
Ventilatory Ratio = VE/(VE ideale) ∙ PaCO2/(PaCO2 ideale).
Come detto sopra il VE ideale è calcolato come 0.1 L/min per kg di peso ideale e la PaCO2 ideale è stimata in 37.5 mmHg.
Nel caso di Mario Rossi, che ha un peso ideale di 70 kg, il Ventilatory Ratio quando ipoventila è:
Ventilatory Ratio = 3 L/min / 7 L/min ∙ 80 mmHg/ 37.5 mmHg = 0.9.
Ciò significa che Mario Rossi necessita di una VE pari alla 0.9 volte la VE ideale per mantenere la PaCO2 a 37.5 mmHg. Valori di Ventilatory Ratio vicini a 1 indicano l’assenza o la scarsa rilevanza di spazio morto: la VE “ideale” è sufficiente a mantenere la normocapnia, come in fisiologia.
Quando invece invece Mario Rossi ha l’ARDS, il Ventilatory ratio è:
Ventilatory Ratio = 12 L/min / 7 L/min ∙ 70 mmHg/37.5 mmHg = 3.2.
In altre parole, in questa condizione Mario Rossi ha bisogno di più del triplo della VE ideale per mantenere la normocapnia, segno di un grave aumento di spazio morto.
Un valore di Ventilatory Ratio superiore a 2 si associa ad incremento del rischio di morte nei pazienti con ARDS.
Conclusioni.
Riassumiamo i punti principali del post:
- L’ipercapnia ha un significato clinico ben diverso se secondaria a riduzione della ventilazione minuto o ad aumento dello spazio morto;
- L’ipercapnia secondaria a riduzione della ventilazione minuto è facile da correggere con la ventilazione meccanica; la correzione dell'ipercapnia secondaria ad aumento dello spazio morto può invece essere pericolosa per il paziente, perchè può portare ad una ventilazione non protettiva;
- La valutazione dello spazio morto è utile nei pazienti con ARDS a fini prognostici e clinici;
- Nella pratica clinica Ventilazione Minuto corretta e Ventilatory Ratio sono dei surrogati appropriati di spazio morto ottenibili in qualsiasi Terapia Intensiva.
Come sempre, un sorriso a tutti gli amici di ventilab.
Bibliografia:
Fisiopatologia dello spazio morto:
- Robertson HT. Dead space: the physiology of wasted ventilation. Eur Respir J 2015;45:1704–1716.
Associazione spazio morto-mortalità nella ARDS:
- Nuckton TJ, Pittet J-F, Kallet R, Daniel BM, Pittet J-F, Eisner M, Matthay MA. Pulmonary Dead-Space Fraction as a Risk Factor for Death in the Acute Respiratory Distress Syndrome. N Engl J Med 2002;346:1281–1286.
- Cepkova M, Kapur V, Ren X, Quinn T, Zhuo H, Foster E, Liu KD, Matthay MA. Pulmonary Dead Space Fraction and Pulmonary Artery Systolic Pressure as Early Predictors of Clinical Outcome in Acute Lung Injury. Chest 2007;132:836–842.
- Kallet RH, Zhuo H, Liu KD, Calfee CS, Matthay MA, on behalf of the National Heart Lung and Blood Institute ARDS Network Investigators. The Association Between Physiologic Dead-Space Fraction and Mortality in Subjects With ARDS Enrolled in a Prospective Multi-Center Clinical Trial. Respiratory Care 2014;59:1611–1618.
Associazione VEcorr/Ventilatory ratio e mortalità nella ARDS:
- Sinha P, Singh S, Hardman JG, Bersten AD, Soni N. Evaluation of the physiological properties of ventilatory ratio in a computational cardiopulmonary model and its clinical application in an acute respiratory distress syndrome population. Br J Anaesth 2014;112:96–101.
- Sinha P, Calfee CS, Beitler JR, Soni N, Ho K, Matthay MA, Kallet RH. Physiologic Analysis and Clinical Performance of the Ventilatory Ratio in Acute Respiratory Distress Syndrome. Am J Respir Crit Care Med 2019;199:333–341.
- Fusina F, Albani F, Bertelli M, Cavallo E, Crisci S, Caserta R, Nguyen M, Grazioli M, Schivalocchi V, Rosano A, Natalini G. Corrected Minute Ventilation Is Associated With Mortality in ARDS Caused by COVID-19. Respir Care 2021;66:619–625.
Spazio morto nella ARDS da COVID-19 e da altre malattie:
- Bertelli M, Fusina F, Prezioso C, Cavallo E, Nencini N, Crisci S, Tansini F, Mazzuca Mari L, Hoxha L, Lombardi F, Natalini G. COVID-19 ARDS is characterized by increased dead space ventilation compared with ARDS from other diseases. A cohort study. Respir Care, in press.