Giorgio ha una ARDS. L’impostazione della ventilazione meccanica è: volume corrente (V
T) 420 ml, frequenza respiratoria (FR) 26/min, PEEP 8 cmH
2O. Il PaO
2/F
IO
2 è 170 mmHg, la PaCO
2 68 mmHg ed il pH 7.36. Dobbiamo preoccuparci di questa ipercapnia?
In passato ho speso parole di tolleranza verso l’incremento della PaCO2 in corso di ventilazione protettiva nei pazienti con ARDS (vedi post del 24/09/2011 e del 03/08/2013). Questa tolleranza è ancora giustificata dopo la pubbblicazione di uno studio (che ultimamente vedo citato sempre più spesso) che evidenzia un’associazione tra ipercapnia e mortalità nei pazienti con ARDS (1)? Questo dato, se credibile, ci deve ovviamente creare forti dubbi se accettare o meno l’ipercapnia di Giorgio.
Lo studio che documenta l’associazione tra ipercapnia e mortalità è un’analisi secondaria su pazienti arruolati in tre studi osservazionali condotti tra il 1998 ed il 2010 ed il livello di ipercapnia è stato definito in base al peggior valore di PaCO2 rilevato nelle prime 48 ore di ventilazione meccanica. Siamo di fronte ad un disegno dello studio certamente “debole“, e perciò i risultati devono essere interpretati criticamente, soprattutto alla luce della plausibilità biologica. Per questo può essere utile, e piacevole, un tuffo nella fisiologia per capire perchè si genera l’ipercapnia nei pazienti con ARDS e quali sono le sue conseguenze biologiche e cliniche.
La pressione parziale della CO2 alveolare (PACO2).
La pressione parziale della CO2 alveolare (PACO2, con la “A” maiuscola per identificare l’alveolo) dipende della produzione di CO2 (V’CO2) e della ventilazione alveolare (VA). In particolare, la PACO2 aumenta se la ventilazione alveolare VA diminuisce. Il tutto è riassunto con semplice efficacia nell’equazione dei gas alveolari per la CO2:
La ventilazione alveolare VA differisce dalla ventilazione minuto VE (il prodotto di VT e FR) perchè esclude la parte di VT che resta confinata nello spazio morto (VD), cioè che si ferma nelle vie aeree prive di strutture alveolari e quindi senza possibilità di scambio gassoso:

L’equazione 2 può essere riscritta, in modo forse più utile per la prosecuzione della lettura, come:

(equazione 3)
dove VD/VT è il rapporto tra spazio morto e volume corrente, variabile con la quale torneremo presto a fare i conti. La ventilazione alveolare VA pertanto diminuisce se si riduce la ventilazione minuto VE e/o se aumenta il VD/VT.
L’equazione 1 può essere può essere riformulata nel seguente modo:

(equazione 4)
Assumiamo da questo momento che la V’CO2 sia costante e torniamo al caso di Giorgio. Se è elevata la PaCO2, (con la “a” minuscola per identificare il sangue arterioso), sarà elevata anche la PACO2, poichè PACO2 e PaCO2 sono considerate sostanzialmente equivalenti.
La VE di Giorgio durante la ventilazione protettiva è di 10.9 l/min (420 ml × 26/min), quasi il doppio di quella fisiologica, che è circa 5.4-6 l/min (450-500 ml × 12/min). Ne consegue che l’ipoventilazione alveolare e quindi l’ipercapnia possono essere spiegate unicamente da un aumento del VD/VT (equazione 4).
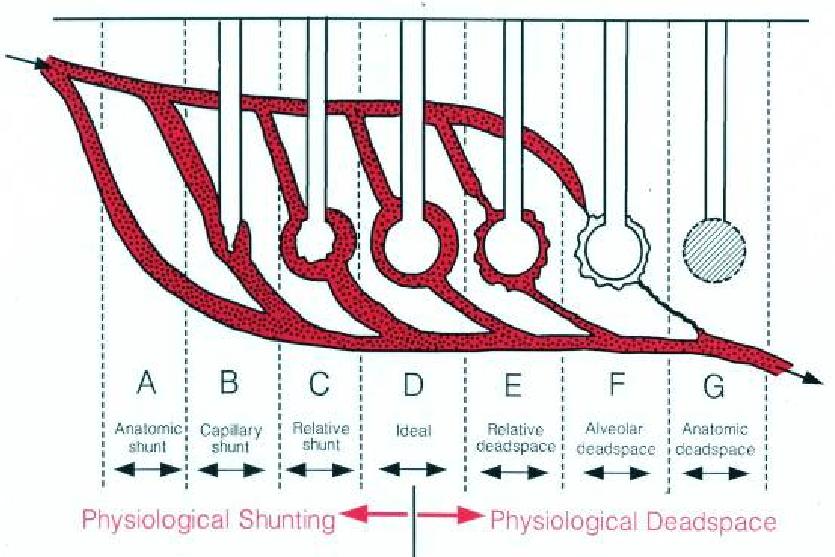
A questo punto complichiamo un po’ il concetto di spazio morto: non parliamo infatti dello spazio morto anatomico (quello delle vie aeree) (VDaw), ma dello spazio morto fisiologico (VDphys), somma del VDaw e dello spazio morto alveolare (VDalv).
Lo spazio morto anatomico VDaw (cioè il volume delle vie aeree) può essere schematizzato come uno spazio aereo anatomicamente sprovvisto di capillari alveolari (G nella figura 1). Il suo volume non si modifica significativamente nella ARDS.
Lo spazio morto alveolare VDalv si genera in quegli alveoli in cui cessa la perfusione capillare, dove quindi la ventilazione non partecipa allo scambio gassoso per l’assenza di contatto con il sangue capillare polmonare (figura 1, F). In queste zone il rapporto ventilazione alveolare/perfusione (VA/Q) è infinito (∞), dal momento che la perfusione è 0.
Nei polmoni patologici esistono anche aree con un eccesso di ventilazione alveolare rispetto alla perfusione (VA/Q elevato, ma non ∞), che contribuiscono allo spazio morto alveolare (figura 1, E). Talvolta questa condizione è definita “spazio morto relativo” o “effetto spazio morto”.
Nella ARDS è l’aumento dello spazio morto alveolare che determina l’incremento dello spazio morto fisiologico.

La figura 2 propone un grafico tradizionale in tutti i testi di fisiologia: la variazione delle pressioni dei gas alveolari al variare del VA/Q. Occupiamoci ora solo della PACO2 (asse verticale): nelle zone con VA/Q = ∞ (i casi F e G della figura 1), la PACO2 è 0 mmHg: l’aria inspirata, che non contiene CO2, non viene modificata per l’assenza di scambio gassoso (figura 2, punto A). La progressiva riduzione del VA/Q (freccia rossa tratteggiata) determina un consensuale aumento della PACO2 (freccia grigia che va da A a B), che al massimo può eguagliare la pressione venosa mista di CO2 (PvCO2) quando il VA/Q diventa 0.

Forse l’argomento non è però così chiaro come può sembrare: l’aumento del VA/Q, che produce VDalv, determina un calo della PACO2 (figura 2). Ma nell’equazione 4 abbiamo visto che l’incremento del VD aumenta la PACO2. Pensiamo alla ARDS, dove coesistono aree con alto e basso VA/Q: nelle zone ad elevato VA/Q, la PACO2 dovrebbe essere bassa, in quelle a basso VA/Q al massimo dovrebbe essere simile a quella venosa (fisiologicamente 46 mmHg) (sempre figura 2). Come possiamo allora spiegare l’ipercapnia di Mario se la PACO2 (e la PaCO2 che è ad essa simile) non raggiunge valori elevati, qualunque sia il VA/Q? Come si può ridurre la VA a tal punto da generare una marcata ipercapnia?
La relazione tra spazio morto e PaCO2 è quindi piuttosto complessa e merita un approfondimento per risolvere le vere o presunte contraddizioni.
La CO2 espirata, alveolare ed arteriosa.
Vedremo ora una descrizione dettagliata dell’impatto che il VA/Q ha su PaCO2 e spazio morto alveolare: la prima parte, relativa alla fisiologia, è fatta passo passo, per consentire poi di muoversi più agilmente negli esempi successvi. Premetto che è una interpretazione originale: ho cercato di seguire una strada diversa da quella che si trova solitamente nei testi ed in letteratura. Ho preferito questo approccio (con i rischi e i limiti che può avere) perchè seguendo l’approccio tradizionale non ho mai capito veramente bene come si possa generare l’ipercapnia quando in assenza di una riduzione della ventilazione al minuto.
Ipotizziamo di avere un soggetto sano, da “libro di fisiologia”: VT 500 ml, VDaw 150 ml, FR 11/min, V’CO2 200 ml/min. Poichè i suoi polmoni sono sani ed ha una fisiologica distribuzione del VA/Q, tutto il suo spazio morto corrisponde a quello anatomico ed è circa il 30% del volume corrente (VD/VT = 150 ml/500 ml = 0.30). La VE è 5500 ml/min (500 ml × 11/min): la VA è 3850 ml/min [(500 ml -150 ml) × 11/min], mentre 1650 ml/min è la ventilazione al minuto dello spazio morto (150 ml × 11/min).
Immaginiamo i polmoni divisi in due compartimenti di identiche dimensioni, in cui la VA sia distribuita al 50% in ciascun compartimento polmonare (cioè 1925 ml/min per compartimento).
La CO2 prodotta ogni minuto dal metabolismo tissutale (cioè il V’CO2) è trasportata ai capillari polmonari. In condizioni di equilibrio, i polmoni eliminano solamente e completamente una quantità di CO2 uguale a quella prodotta dai tessuti e non più di questa. Prendiamo in considerazione questa condizione di stabilità, in cui il volume di CO2 espirato coincida perfettamente con la produzione di CO2.
Consideriamo la portata cardiaca Q distribuita equamente nei due compartimenti. Poichè la CO2 prodotta dal metabolismo è portata ai capillari alveolari dalla portata cardiaca, vi sarà anche una uguale ripartizione del V’CO2 nei due comportimenti, a ciascuno dei quali saranno indirizzati quindi 100 ml di CO2 ogni minuto (cioè il 50% del V’CO2). E’ questa la quantità di CO2 che ciascun compartimento deve eliminare all’equilibrio attraverso la ventilazione alveolare.
Questa condizione è schematizzata nella figura 3. I due compartimenti sono delimitati nelle aree azzurra e rosa. L’apparato respiratorio, delimitato dalla linea nera continua, è composto da una “Y” capovolta (le vie aeree, cioè il VD), esterna ai due compartimenti polmonari, e da due spazi alveolari circolari, ciascuno in un differente compartimento. La doppia riga continua rossa schematizza i rispettivi capillari polmonari, nel cui interno è quantificato il flusso di CO2 che, trasportato dal sangue, deve essere “smaltito” nei polmoni ogni minuto.
Ed ora veniamo al dunque.
Ci troviamo nella condizione in cui, in ciascun compartimento, 100 ml di CO2 si miscelano in 1925 ml di VA. Possiamo quindi calcolare la frazione alveolare di CO2 (FACO2), cioè il rapporto tra il volume di CO2 ed il volume alveolare (per la precisone è, in questo caso, un rapporto tra flussi). La FACO2 in questo caso è 0.052 (cioè 100 ml/min di CO2 in 1925 ml/min di VA): questo significa che la CO2 rappresenta il 5.2% del gas alveolare (FACO2 × 100). Per la legge delle pressioni parziali dei gas di Dalton, la PACO2 esercita il 5.2% della pressione presente nell’alveolo, che pertanto è 40 mmHg (760 mmHg × 0.052) (Figura 4). (Ipotizziamo per semplicità un quoziente respiratorio di 1, che significa che 100 ml di CO2 che entrano nell’alveolo sostituiscono una uguale quantità di O2 che lascia l’alveolo per entrare nel sangue capillare).

La pressione di CO2 nel gas espirato (PECO2) non è uguale alla PACO2, perchè il gas alveolare si deve miscelare con quello dello spazio morto, che non contiene CO2. Il modo più semplice per conoscere l’ipotetica PECO2 è calcolare la frazione espiratoria di CO2 (FECO2) e moltiplicarla per la pressione atmosferica (similmente a quanto abbiamo fatto per il calcolo della PACO2). E’ probabilmente più facile capire il concetto seguendo i calcoli: all’uscita dalle vie aeree, i 200 ml di CO2, prodotti ogni minuto dal metabolismo ed espirati, sono contenuti nel VE (che nell’esempio è 5500 ml/min); la FECO2 è 0.04 (200/5500) e la PECO2 28 mmHg (760 x 0.04) (Figura 5).

Resta da capire quanto sarà, in questa condizione, la PaCO2. In ciascuno dei compartimenti, la PACO2 si mette in equilibrio con il rispettivo sangue capillare, e la pressione parziale di CO2 alla fine del capillare polmonare (PcCO2) diventa uguale alla corrispondente PACO2. In entrambi i compartimenti la PcCO2 è quindi 40 mmHg. Poichè la portata cardiaca è ripartita equamente, il flusso di sangue in uscita dal circolo capillare polmonare è uguale nei due compartimenti. La PaCO2 è la media ponderata delle PcCO2 dei due compartimenti (cioè la somma dei prodotti tra PcCO2 e frazione di Q in ciascun compartimento); in questo caso è facile: 40 mmHg (cioè: 40 × 0.5 + 40 × 0.5) (Figura 6). (Può essere ovvio, ma è meglio ricordare che la frazione di Q è la percentuale di Q/100)
Ora possiamo calcolare il rapporto tra spazio morto fisiologico e volume corrente con la formula di Bohr-Enghoff:

C’è dibattito se questo approccio sia preferibile alla originale formula di Bohr (calcolabile con la capnografia volumetrica), dove la PACO2 sostituisce la PaCO2. Se qualcuno fosse interessato, potremmo discuterne nei commenti.
Il VDphys/VT che calcoliamo con i nostri dati è 0.30, uguale allo VDaw/VT, calcolato con lo spazio morto anatomico della nostra simulazione. Ne deduciamo che in questa condizione non esiste spazio morto alveolare perche spazio morto fisiologico ed anatomico coincidono.
Ipotizzando una portata cardiaca da fisiologia (5000 ml/min), il VA/Q complessivo sarebbe 0.8, anch’esso ovviamente fisiologico.
Ventilazione protettiva, ARDS e ipercapnia.
Applicando la ventilazione protettiva di Giorgio (VT 420 ml × FR 26/min) ad un polmone fisiologico, come quello esaminato nell’esempio precedente, quanto sarebbe la PaCO2?
Vediamo il risultato finale nella figura 7.
Risparmiamoci tutti i passaggi visti nell’esempio precedente (è comunque un ottimo esercizio per chi lo volesse fare) e arriviamo al risultato finale: la ventilazione protettiva applicata ad un soggetto sano produce ipocapnia (nel nostro esempio la PaCO2 è 22 mmHg). Questa ventilazione sarà anche protettiva, ma è pur sempre una forma di iperventilazione.
Rispetto alla ventilazione fisiologica, il VDphys/VT è un po’ aumentato (da 0.30 a 0.36), ma come nell’esempio di prima è uguale al VDaw/VT (150 ml di VDaw su 420 ml di VT): anche in questo caso non vi è spazio morto alveolare, dato che non vi è differenza tra VDphys/VT e VDaw/VT . L’aumento della ventilazione alveolare (da 3850 a 7020 ml/min) è stato molto maggiore dell’aumento di spazio morto, e questo spiega la riduzione della PACO2 e conseguentemente della PaCO2.
Se la portata cardiaca rimanesse uguale a quella dell’esempio precedente (5000 ml/min), il VA/Q complessivo sarebbe 1.4, un mismatch ventilazione/perfusione con alto VA/Q.
Facciamo ora l’esempio di un paziente con ARDS, come Giorgio, a cui viene erogata la ventilazione protettiva con VT 420 ml e FR 26/min. Il polmone è certamente diverso da quello fisiologico.

Figura 8
Nella figura 8 vediamo una sezione TC di polmone sano a sinistra e con ARDS a destra. Nel polmone sano vediamo che il parenchima polmonare è di colore omogeneo. Nel polmone con ARDS vediamo aree iperdiafane (più nere del normale, delimitate dalla linea azzurra) ed aree iperdense (più bianche del normale, linea rossa). Nelle zone iperdiafane vi è sovradistensione delle strutture alveolari, dovuta ad un incremento della ventilazione regionale. Vi è anche una riduzione della perfusione, in parte per un effetto gravitazionale, in parte perchè le pareti alveolari iperdistese comprimono i capillari polmonari. Nelle aree iperdense vi è una riduzione (o l’assenza) di ventilazione, con la perfusione che è favorita dalla forza di gravità, con un effetto variabile sulle resistenze vascolari legato all’entità della vasocostrizione ipossica.
Ipotizziamo che nelle zone più ventilate venga dirottato il 80% delle ventilazione, mentre in quelle meno ventilate sia distribuito il rimanente 20%. Ed ipotizziamo che la perfusione sia distribuita prevalentemente (80%) alle zone poco ventilate ed in piccola parte alle zone iperventilate (20%). La condizione è schematizzata nella figura 9, in cui nel compartimento di sinistra (azzurro) sono schematizzate le aree ben ventilate e mal perfuse ed in quello di destra (rosso) quelle mal ventilate e ben perfuse.
Figura 9
Abbiamo quindi un compartimento ad alto VA/Q (a sinistra) ed una a basso VA/Q (a destra). A sinistra destra arrivano 5616 ml/min (cioè l’80%) di ventilazione che devono drenare, all’equilibrio, il 20% della CO2 prodotta dall’organismo, dal momento che vi arriva solo il 20% della portata cardiaca. A destra sinistra una ventilazione alveolare molto minore (1404 ml/min, cioè il 20 %) deve farsi carico del 80% del V’CO2. Il risultato finale è mostrato in figura 10.

Come sempre, puoi rifare i calcoli da solo (il procedimento è sempre lo stesso). La sostanza è che, nel compartimento di sinistra, la rimozione di poca CO2 da parte di tanta ventilazione porta ad un drastico abbassamento della PACO2 e quindi della PcCO2. Viceversa, nel compartimento di destra, il passaggio di tanta CO2 in poco volume ventilato è possibile solo raggiungendo una elevata PACO2 e quindi una altrettanto elevata PcCO2. La PaCO2 è la media ponderata del sangue proveniente dai capillari polmonari: 5 mmHg*0.2 + 87 mmHg*0.8 = 70 mmHg. Il risultato finale è una PaCO2 elevata (70 mmHg, più o meno come quella di Giorgio), frutto del fatto che molto sangue con 87 mmHg di PCO2 si miscela a poco sangue con 5 mmHg di PCO2.
Il VDphys/VT è di 0.80, ben più elevato dello 0.36 del VDaw/VT (150 ml di VDaw/420 ml di VT). In questo caso abbiamo un notevole spazio morto alveolare, essendo il VDalv/VT 0.44 (cioè la differenza tra VDphys/VT e VDaw/VT). Se applichiamo l’equazione 4, si calcola una VA di 2184 ml/min, di gran lunga inferiore ai 7020 ml/min ottenuti utilizzando il solo spazio morto anatomico. Ed anche inferiore ai 3850 ml/min della condizione fisiologica che è stata considerata inizialmente. A questo punto si potrebbero fare molte speculazioni sul significato di ventilazione alveolare e spazio morto fisiologico… certamente ciascuno le potrà fare per proprio conto…
I calcoli che abbiamo fatto non hanno certo la pretesa di essere precisi in vivo e ci sono aspetti complessi di cui non si è tenuto conto. Ma fanno capire bene che l’ipercapnia si genera nelle zone a basso VA/Q: è sempre l’ipoventilazione alveolare a determinare l’aumento della PACO2 e della PaCO2. In alcuni casi il concetto è chiaro, come quando vi è ipoventilazione (riduzione della VE), che ha come inevitabile conseguenza un’omogenea riduzione della VA e quindi del VA/Q. Quando invece si ha iperventilazione (elevata VE) e nonostante questo si sviluppa ipercapnia, essa è generata dal mismatch VA/Q che si genera nelle zone a basso VA/Q regionale, dove si instaura una ipoventilazione distrettuale perchè altre aree polmonari hanno “rubato” ventilazione e ceduto perfusione.
Può ora essere evidente come ipossiemia ed ipercapnia si sviluppino negli stessi compartimenti polmonari, quelli in cui il VA/Q regionale è inferiore a quello fisiologico: poca ventilazione modifica poco il sangue venoso, e l’elevata perfusione fa sì che queste zone abbiano un peso più rilevante sulla composizione del sangue arterioso. Viceversa i compartimenti con alto VA/Q regionale influiscono poco sui gas arteriosi: sono zone a relativamente bassa perfusione e quindi contribuiscono meno alla composizione del sangue arterioso.
Per concludere (per ora)…
Un recente articolo ci ha fatto venire il dubbio che l’ipercapnia possa essere “velenosa” per i pazienti con ARDS, avendo trovato un’associazione tra ipercapnia e mortalità.
Abbiamo capito che le alterazioni regionali del VA/Q sono tipiche dei pazienti con ARDS (figura 8) e che sono la causa dell’ipercapnia durante la ventilazione protettiva. Il mismatch VA/Q è correlato sia all’estensione degli addensamenti polmonari che alle alterazioni della perfusione polmonare. In sostanza, più è grave il paziente con ARDS (sommando le disfunzioni polmonare e cardiocircolatoria), più aumenta il mismatch VA/Q, più aumenta la PaCO2.
L’ipercapnia è quindi un ottimo marker della gravità del paziente con ARDS.
Questo non esclude però che l’ipercapnia possa avere anche un effetto negativo diretto sulle funzioni dell’organismo. Il post di oggi è già lunghissimo, nel prossimo cercheremo di capire gli effetti biologici dell’ipercapnia, per poter finalmente discutere appropriatamente i risultati dello studio da cui abbiamo preso le mosse.
Un sorriso a tutti gli amici di ventilab.
Bibliografia.
1) Nin N et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med 2017;43:200-8